Informationen zur europäischen Medical Device Regulation
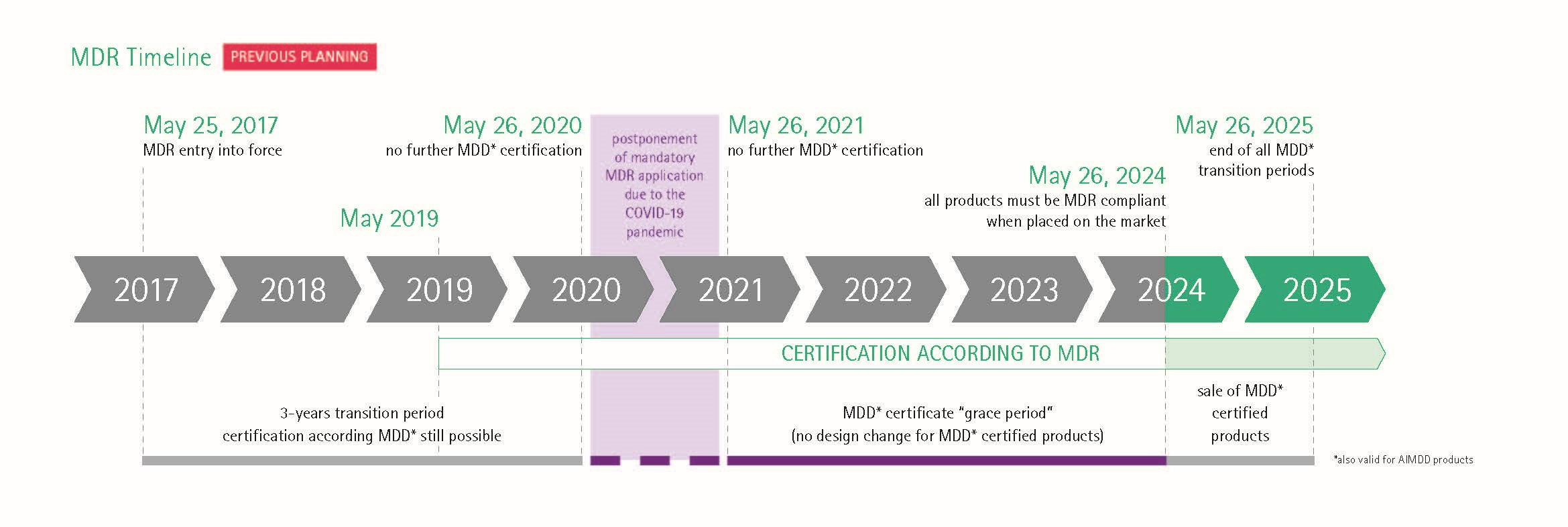
Im Mai 2020 sollte die Übergangsfrist für die neue europäische Medizinprodukteverordnung mit vielen neuen Regelungen und Herausforderungen für alle Beteiligten enden. Die Übergangsfrist wurde aufgrund der Corona-Pandemie um 1 Jahr verlängert. Neuer Geltungsbeginn ist Mai 2021.
B. Braun bereitet sich intensiv vor und will die neuen Anforderungen so schnell wie möglich umsetzen. Hier haben wir einige Informationen über die MDR für Sie zusammengestellt.
Die neue europäische Medizinprodukteverordnung (Medical Device Regulation, MDR) trat im Mai 2017 in Kraft. Die neue Verordnung löst die bestehende Medizinprodukte-Richtlinie MDD (Medical Device Directive) und die AIMDD (Active Implantable Medical Device Directive) ab. Bis zum Ablauf einer Übergangsphase im Mai 2021 dürfen Medizinprodukte aber auch weiter nach den bisherigen Richtlinien zertifiziert werden. Für die IVDR (In-vitro-Diagnostika Regulation), die die IVDD (In-vitro-Diagnostika Directive) ablöst, gilt eine abweichende Übergangsfrist von 5 Jahren – bis Mai 2022, die unter bestimmten Voraussetzungen sogar um 2 Jahre verlängert werden kann.
Zurzeit existieren ca. 500.000 Medizinprodukte in Europa, die von der MDR betroffen sind. Schätzungen zufolge werden aber nur ungefähr 65 Prozent der Medizinprodukte gemäß dem neuen, wesentlich umfangreicheren Regelwerk zum Erhalt des „CE-Zeichens“ (re-)zertifiziert werden. Aktuell befindet sich der überwiegende Anteil der Benannten Stellen in der Phase der Akkreditierung. Es ist noch offen, wie viele Prüfstellen zu welchem Zeitpunkt das Verfahren abschließen können (aktuelle Liste der Europäischen Kommission). Aufgrund der gestiegenen Anforderungen an die Benannten Stellen und die Hersteller sind Portfolio-Anpassungen zu erwarten und unumgänglich.
Hintergrund
Welche Regelungsbereiche sind betroffen?
Unter anderem sind diese Bereiche betroffen:
Klassifizierungsregeln:
- Die Regeln sind um die Klasse Ir (wiederverwendbare chirurgisch-invasive Instrumente) erweitert worden.
- Die Anforderungen an implantierbare Produkte der Klasse IIb sind gestiegen.
- Es wurden neue Risikoklassen für einige Produktkategorien eingeführt, wodurch Produkte ggf. höher eingestuft werden.
Klinische Evidenz:
- Künftig unterliegen Medizinprodukte aller Klassen der Pflicht zur klinischen Bewertung.
Scrutiny-Verfahren:
- Neue implantierbare Produkte der Klasse III und medikamentenfreisetzende Produkte der Klasse IIb werden vor Markteintritt stärker kontrolliert.
Benannte Stellen (Notified Bodies):
- Die Anforderungen an die Benannten Stellen steigen. Gleichzeitig sind sie künftig verpflichtet, unangekündigte Audits bei Herstellern durchzuführen.
Technische Dokumentation:
- Der Umfang der Dokumentation steigt durch die MDR erheblich, wodurch auch der Aufwand für die Hersteller deutlich zunimmt.
EUDAMED:
- Einführung einer elektronischen Datenbank zur Überwachung des Produktlebenszyklus.
Wo liegen die Herausforderungen?
Um zukünftig die kontinuierliche Versorgung mit sicherer und innovativer Medizintechnik gewährleisten zu können, stehen alle Hersteller vor der Herausforderung, die gestiegenen Anforderungen zum Erhalt des CE-Zeichens zu bewältigen. Die Benannten Stellen müssen die Voraussetzungen und ausreichende Kapazitäten für das Konformitätsbewertungsverfahren schaffen. Ob dies innerhalb des bekannten, vorgegebenen Zeitrahmens zur Implementierung der MDR realisiert werden kann, kann gegenwärtig noch nicht beantwortet werden.
B. Braun hat bereits zu einem frühen Zeitpunkt mit umfangreichen Vorbereitungen für die Zertifizierung der eigenen Medizinprodukte gemäß MDR begonnen. Das gilt selbstverständlich für alle B. Braun Artikel, sowohl von B. Braun selbst hergestellte Produkte als auch auf dem Markt zur Komplettierung des Portfolios eingekaufte Waren. Im Hinblick auf den Fortschritt der getroffenen Maßnahmen ist B. Braun zuversichtlich, die Anforderungen der MDR erfüllen zu können.

FAQs
Allgemeine FAQs zum Thema MDR
Was sind die Gründe für die neue Medizinprodukte-Verordnung?
Für die 1993 veröffentlichte und bis heute noch gültige Medizinprodukte-Richtlinie 93/42 EEC (MDD) wurde eine Überarbeitung auf europäischer Ebene erforderlich.
Mit der neuen Verordnung möchte die EU-Behörde die Qualität und Sicherheit von Medizinprodukten verbessern, die Prozesse EU-weit harmonisieren und die Patientensicherheit erhöhen.
Weitere Aspekte sind die Verbesserung der Transparenz und Rückverfolgbarkeit in Verbindung mit neuen Technologien, die eine eindeutige Identifikation aller Produkte über die gesamte Lebensdauer ermöglichen.
Was ist in der Medizinprodukte-Verordnung (MDR) geregelt?
Die MDR definiert die Anforderungen, die ein Hersteller erfüllen muss, um Medizinprodukte in Europa zu verkaufen. Betroffen sind sowohl die technischen Anforderungen an ein Produkt als auch Anforderungen an die Beobachtung von Produkten, die in Einrichtungen des Gesundheitswesens eingesetzt werden.
Im Unterschied zur vorherigen Regelung (MDD) handelt es sich um eine europäische Verordnung, die direkt in allen europäischen Ländern gilt.
Aufgrund der COVID-19-Pandemie wurde die Übergangsfrist für die obligatorische Anwendung der MDR bis Mai 2021 verlängert.
Welche wesentlichen Änderungen ergeben sich durch die Medizinprodukte-Verordnung (MDR)?
Zur Klassifizierung von Produkten gibt es mehrere Veränderungen. Zusätzlich zur Einführung der neuen Klasse Ir für wiederverwendbare chirurgische Instrumente, haben sich besonders die Anforderungen an implantierbare Produkte der Klasse IIb erhöht. Darüber hinaus wurden zahlreiche Produktkategorien einer höheren Risikoklasse zugeordnet.
Die MDR erhöht die Anforderungen an die klinische Evidenz von Medizinprodukten. Zukünftig erfordern alle Medizinprodukte, unabhängig von ihrer Risikoklasse, eine klinische Bewertung. Das neu eingeführte „Scrutiny“-Verfahren bedeutet eine verbesserte Überwachung von neuen, implantierbaren Produkten der Risikoklasse III, sowie von Arzneimittel-haltigen Produkten der Klasse IIb vor Markteintritt.
Zusätzlich zu den erhöhten Anforderungen an den Hersteller gelten ab sofort auch strengere Regeln für die Benannten Stellen. Um Medizinprodukte zulassen zu dürfen, müssen diverse zusätzliche Anforderungen erfüllt werden. Zudem sind die Benannten Stellen dazu verpflichtet, unangekündigte Audits bei den Herstellern durchzuführen. Zusätzliche Anforderungen an die von den Herstellern zu liefernde technische Dokumentation erhöhen den Umfang und die Komplexität der Dokumentation erheblich.
Was ist die EUDAMED?
Für die Öffentlichkeit relevante Daten werden in einer schon heute existierenden, zentralen europäischen Datenbank zugänglich gemacht. Die erweiterte Version der EUDAMED wird bis Mitte 2022 eingeführt.
In der EUDAMED müssen Hersteller, Importeure oder für den Vertrieb in der EU autorisierte Vertreter für Medizinprodukte Daten zu ihrer Rolle als Wirtschaftsakteure, sowie produktrelevante Daten über jedes, für den Vertrieb in der EU bestimmte Produkt zur Verfügung stellen.
Betrifft die MDR alle Produkte?
Ja, alle Medizinprodukte der verschiedenen Risikoklassen inklusive Behandlungseinheiten und Systeme sind betroffen.
In-vitro-Diagnostika, deren Inverkehrbringen in der neuen In-vitro-Diagnostic Device Regulation (IVDR) geregelt sind, sind ebenfalls betroffen.
Wie ist der Zeitplan der MDR-Einführung?
Nach Veröffentlichung der MDR am 5. Mai 2017 trat die Verordnung am 25. Mai 2017 mit einer Übergangsphase, die bis zum 26. Mai 2020 dauern sollte, in Kraft.
Aufgrund der COVID-19-Pandemie wurde die obligatorische Anwendung der MDR auf den Mai 2021 verschoben.
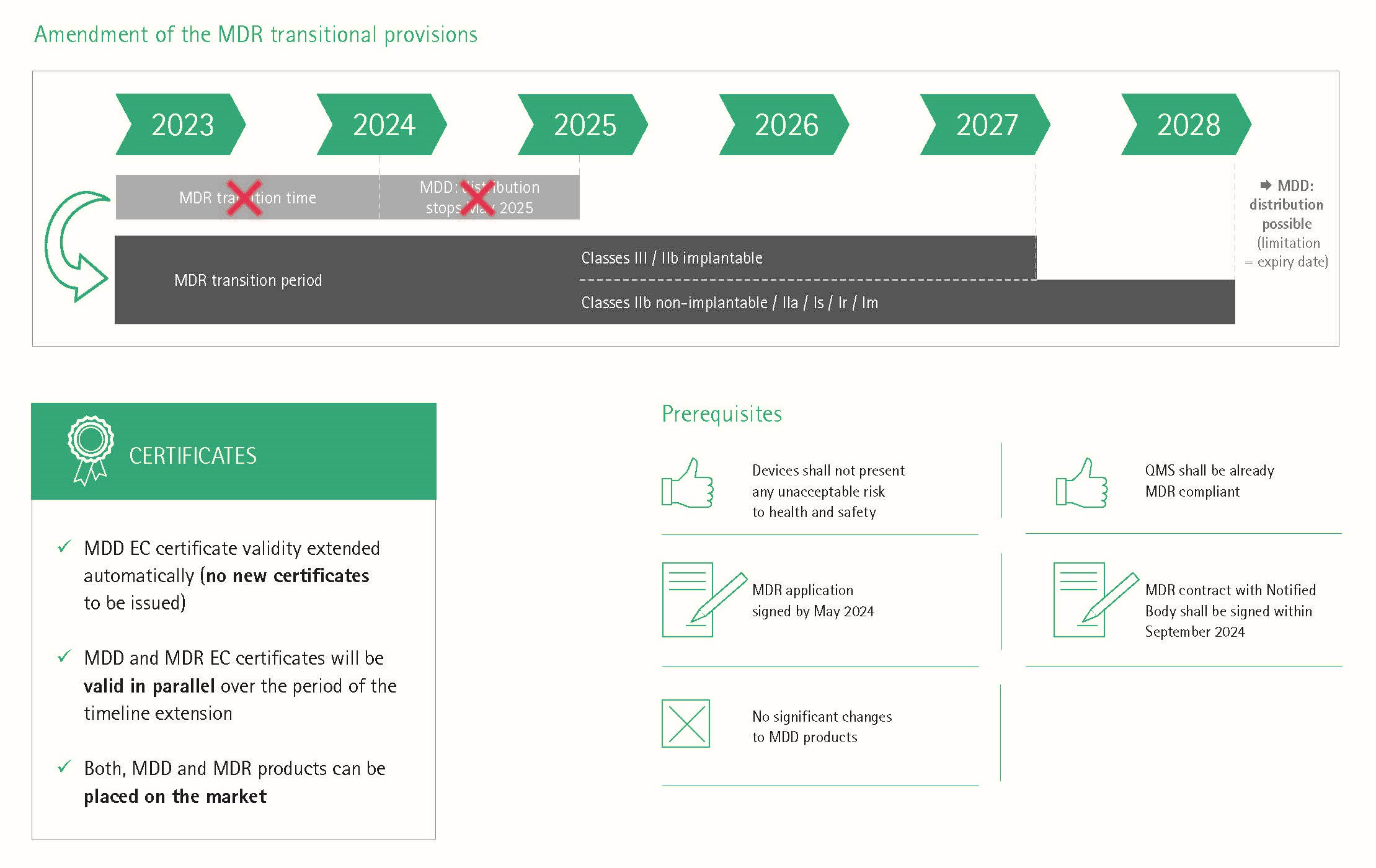
Bis zum 26. Mai 2024 behalten MDD-Zertifikate noch ihre Gültigkeit (z.B. für Produkte der Risikoklassen II und III), es sei denn, es ist vorgeschrieben, das MDD-Zertifikat durch ein MDR-Zertifikat zu ersetzen (z.B. für Produkte der Risikoklasse I).
Nach dem 26. Mai 2025 dürfen Produkte mit einem MDD-Zertifikat nicht mehr auf den Markt gebracht werden.
Welche MDR Klassen haben ab wann ihren Geltungsbeginn?
Aufgrund der COVID-19 Pandemie wurde die verbindliche Anwendung der MDR auf den 26. Mai 2021 festgelegt. Damit ergeben sich folgende Deadlines für das Inverkehrbringen von Medizinprodukte nach Produktklassen:
§ Klasse I: 26. Mai 2021
§ Klasse Ir, s, m, Klasse IIa, Klasse IIb und Klasse III: 26. Mai 2024
Was ist die Konformitätsbewertung?
Die Konformitätsbewertung sagt aus, ob ein Produkt und der jeweilige Hersteller die europäischen MDR-Anforderungen erfüllen. Abhängig von der Risikoklassifizierung der einzelnen Produkte ist B. Braun dazu berechtigt, diese Prüfung selber durchzuführen.
Die Bewertungen für höhere Klassifikationen (Klassen Ix, IIa, IIb, III) erfolgen durch eine sogenannte „Benannte Stelle“. Wie zuvor gibt es drei Risikoklassen. Eine neue Sub-Risikoklasse „Ir“ für wiederverwendbare chirurgische Instrumente wurde eingeführt. Die Konformitätserklärung ist Basis für die CE-Kennzeichnung.
Was ist eine Benannte Stelle?
Eine Benannte Stelle ist ein privates Unternehmen, das nach Akkreditierung, im Auftrag der Europäischen Union „benannt“ (d.h. beauftragt) wird, die Konformität eines Herstellers mit der MDR zu überprüfen. Die Benannten Stellen der B. Braun Melsungen AG sind z.B. TÜV SÜD, MedCert, Dekra.
Welche Kennzeichnung bekommen MDR-zertifizierte Produkte?
Mit der Einführung der MDR wird ein neues Symbol "MD" für Medizinprodukte (Medical Devices) eingeführt, das auf die Etiketten aufgebracht wird.
Welcher zusätzliche Aufwand bleibt nach Ablauf der Übergangsfrist am 26. Mai 2024?
Auch nach der Umstellung des gesamten Produktportfolios auf die MDR kommen auf die Hersteller durch die erhöhten Anforderungen der MDR erhebliche Mehrkosten zu.
Welchen Mehrwert bietet die MDR für Hersteller, Gesundheitsdienstleister und Patienten?
Die Hauptziele der Verordnung sind ein besserer Schutz der öffentlichen Gesundheit und der Patientensicherheit, mehr Transparenz, Rechtssicherheit und ein stärker europäisch ausgerichtetes Konzept. Dies soll durch eine umfangreichere technische Dokumentation zu den betroffenen Produkten in einem MDR-konformen Qualitätsmanagementsystem erreicht werden.
FAQs zu MDR & B. Braun
Inwieweit ist B. Braun von der MDR betroffen?
Als Hersteller von Medizinprodukten muss B. Braun die gestellten Anforderungen bis Mai 2021 erfüllen. Verschiedene Arbeitsgruppen aktualisieren die technische Dokumentation und überarbeiten Prozesse, um diese MDR-konform zu gestalten.
Darüber hinaus ist B. Braun dazu verpflichtet, EUDAMED Produktinformationen, insbesondere Produktidentifizierungsdaten (UDI), sowie Post Market Surveillance Informationen zur Verfügung zu stellen.
Welche Produkte von B. Braun sind betroffen?
Alle Medizinprodukte und In-vitro-Diagnostika. Darüber hinaus alle Produkte, die erstmalig gemäss der neuen Klasse Ir zertifiziert werden.
Wird B. Braun die vorgegebene Zeitplanung einhalten können?
Schon seit geraumer Zeit arbeitet B. Braun daraufhin, die Konformität ihrer Produkte mit den neuen Regelungen sicherzustellen und den vorgesehenen Zeitplan einhalten zu können.
Werden alle Medizinprodukte von B. Braun nach MDR zertifiziert werden?
Abhängig vom geplanten Lebenszyklus werden die Produkte nach MDR zertifiziert werden. Nach Ablauf der vorgesehenen Übergangsperiode, d.h. bis spätestens Mai 2024, wird das gesamte Portfolio von B. Braun MDR konform sein.
Innerhalb der Übergangsfrist wird B. Braun sowohl MDD- als auch MDR-zertifizierte Produkte in den Verkehr bringen.
Erfüllen die von B. Braun beauftragten Benannten Stellen die zukünftigen Anforderungen?
Im Mai 2019, wurde der TÜV Süd die zweite Organisation, die weltweit als Benannte Stelle akkreditiert wurde. Weitere Benannte Stellen, die Medizinprodukte von B. Braun kontrollieren, haben die Benennung nach MDR bereits erhalten oder befinden sich in der Akkreditierungsphase. Einen Überblick der nach MDR Benannten Stellen gibt die Webseite der Europäischen Kommission.
FAQs zu MDR & B. Braun Medical Schweiz
Informationen zum MRA (Mutual Recognition Agreement)
Die EU-Medizinprodukteverordnung (MDR) wird am 26. Mai 2021 in Kraft treten. Um den Status quo im Importverfahren für Medizinprodukte von der EU in die Schweiz zu sichern, müsste das Abkommen über die gegenseitige Anerkennung von Konformitätsbewertungen (MRA) zwischen der Europäischen Union (EU) und der Schweiz (CH) vor dem 26. Mai 2021 überarbeitet werden.
Der Ausgang dieses politischen Prozesses ist ungewiss. Wahrscheinlich wird das MRA nicht vor dem 26. Mai 2021 überarbeitet werden und eine Anpassung der Medizinprodukteverordnung zur Klärung der Regelungen für Importe nach dem 26. Mai 2021 wird daher erforderlich sein.
Um Engpässe bei den aus der EU importierten Medizinprodukten von B. Braun aufgrund dieser Rechtsunsicherheit zu vermeiden, hat die B. Braun Medical AG ein Szenario erarbeitet, in welchem die B. Braun Medical AG Schweiz die Funktion der Bevollmächtigten für die Schweiz übernimmt. Dieses Szenario wird bei Bedarf umgesetzt und an die aktualisierte MepV (voraussichtlich am 19 Mai 2021) angepasst.
Dank dieser Massnahme können wir bestätigen, dass alle Produkte von B. Braun auch nach dem 26. Mai 2021 auf dem Schweizer Markt verfügbar sein werden.